
数据科学和机器学习(ML)在催化领域早有应用,但当时预测算法仅被用于建立各种催化反应的定量构效关系(QSPR)。直到最近50年,催化界才更广泛地采用数据驱动方法(图1a)用于研究。而在最近十年间,催化研究广泛利用数据科学概念(数据科学通过一系列分析技术(如统计推断、可视化等)为理解数据提供了基础和背景)来助力催化研究进程。
近期,苏黎世联邦理工学院Javier Pérez-Ramírez等人全面回顾催化研究者如何利用数据驱动策略来解决异相、均相和酶催化的复杂挑战,强调了该领域的前沿以及催化子学科之间的知识迁移,揭示了催化实验在数据探索方面的差距,指出数据科学的四大支柱(描述性、预测性、因果性和规范性分析)能弥补这一差距。最后,作者提倡在实验中采用数据科学方法和数据标准化来促进数字催化的研究。
研究亮点
1、作者对2013-2023年期间的同质、异质和酶催化进行了系统的文献检索,并以数据驱动策略解决催化相关问题的不同类型,最后,作者将所有研究分为演绎型或归纳型。
2、作者提倡催化研究人员对数据驱动概念和策略要有基本了解,但数据驱动不会取代人类的直觉或专业知识。在可预见的未来,数据科学将集成到催化研究中,加速实验设计、数据分析和新知识的创造。
图文导读
通过总结众多的文献报道,作者确定了催化研究中数据驱动方法的两个主要目标。一种是通过绘制结构-性质-性能关系图来验证假设,实现演绎研究。第二种方法需要确定描述符,以便通过可解释的ML模型阐明性能。这些研究的重点是通过揭示性能与性质或性质与结构之间的关系来建立新的理论,称之为归纳研究(图1b)。这些系统性的文献分析以及将催化反应归类为演绎(或归纳方法)的通用模式,为研究提供了一个全新的平台,让人们能够深入了解数据驱动催化研究的全貌(图1c)。

鉴于演绎研究的重要性,作者通过研究数据来源(实验、模拟、文献或数据库)和任务目标(预测、筛选或优化;图2a)等细节分析了它们的范围。在异相催化方面,ML主要用于指导密度泛函理论(DFT)计算(约占65%,主要预测吸附能和电催化反应)。在均相催化方面,ML大部分是实验研究(>60%)。这些研究的目标是筛选最佳底物-溶剂组合等,使用的方法是将催化描述符和工艺参数映射到反应产率和选择性上。
值得注意的是,酶催化展现了理论与实验工作的统一,例如,从现有数据库中检索不同酶的氨基酸序列,然后通过实验室工程实现所需的特性,但大多数研究的主要重点在于预测酶的结构或特性,很少应用于化学反应。尽管如此,这些研究对于推动催化转化用酶的ML辅助定向设计是不可或缺的。接着,作者对异相催化进行了类似的分析,根据驱动力(图2b)对数据进行了分类,观察到热力学性质多于电催化,光催化应用较少。值得注意的是,DFT模拟的数据和文献提取的数据占绝大多数,其目标仅限于理论研究。这表明在异相催化实验中对ML和数据科学的探索还不够。

考虑到催化是化学的分支学科,起源于经验科学,数据收集和分析是理解催化现象并最终促成发现的基础。因此人们致力于开发数据库,如Open Catalysts Dataset、CatHub、ioChem-BD、Open Reaction Database、Kraken、RetroBioCat等,这些数据库收录了有关催化剂结构、性质和性能的信息(图3),推动了数据驱动催化研究的加速发展。

基于数据库,作者强调了“ML算法的选择取决于具体任务和数据可用性”。例如基于树的算法是各学科中最常见选择(占34%),远超了人工神经网络21%的占比(图4)。出现这种趋势的原因是,催化数据通常以数值和表格形式表示,而且往往属于小数据体系,基于树的模型,如随机森林等,在处理此类数据集时与神经网络相比,其更有竞争力或具有更优越的性能。 在图4(a)-(c)中,均相催化领域,线性回归固有的表达式为模型提供了更多可解释性。在酶催化方面,高斯回归等算法更适合处理大数据,通常用于探索氨基酸序列。

随着人工智能和ML的不断进步,将数据驱动方法融入催化研究的前景一片光明。 (1)基于GPT-3.5等语言模型有望在加速化学和催化研究方面发挥关键作用。它们先进的自然语言处理(NLP)和文本挖掘能力有助于从科学文献中高效导航和提取相关信息(图5a)。这些模型通过自动提取结构的合成手段或材料特性数据,加快了文献综述的速度,达到了节省时间和创建化学知识库的双重目的。
(2)主动学习在指导实验、帮助发现和开发高效催化剂方面大有可为。这种方法通过巧妙地平衡探索与开发的概念,缩小潜在反应途径和优化条件的范围,从而简化了化学和参数空间搜索(图5b)。
(3)生成模型(如变分自编码器(VAE)、生成对抗网络(GAN)等)可以加快对未知区域的探索,并创造出具有所需特性的假想分子或材料(图5c)。虽然它们在化学中的应用还处于起步阶段,但VAE和GAN在预测和合成具有目标功能的酶序列方面的成功应用。除了从性质到结构的应用外,它们还可通过从大型反应数据集中学习来预测反应结果,从而提出以前未探索过的反应条件或反应途径。
(4)深度强化学习(DRL)可促进自主学习和决策,减少了对ab initio计算的需求,并加快了最优反应途径或催化剂结构(表面)的发现(图5d)。此外,DRL可以处理多目标问题(同时优化产率和选择性)。

虽然前景一片光明,但数据驱动催化的应用还处于起步阶段,作者认为人工智能的发展将促使这一新兴领域取得更大进展。然而,这需要在实验催化中更多地采用数据科学方法,但目前的趋势并不明显,造成这种差异的因素主要包括:两个领域之间的知识差距,ML只能在大数据(大于1000个数据点)中发挥良好作用的观念,以及对数据驱动模型的黑箱性质缺乏信任。为了解决这些问题,作者详细阐述了描述性分析、预测性分析、因果分析和描述性分析的概念(图6),希望数据驱动工具和策略在实验催化领域得到更广泛的普及和接受。

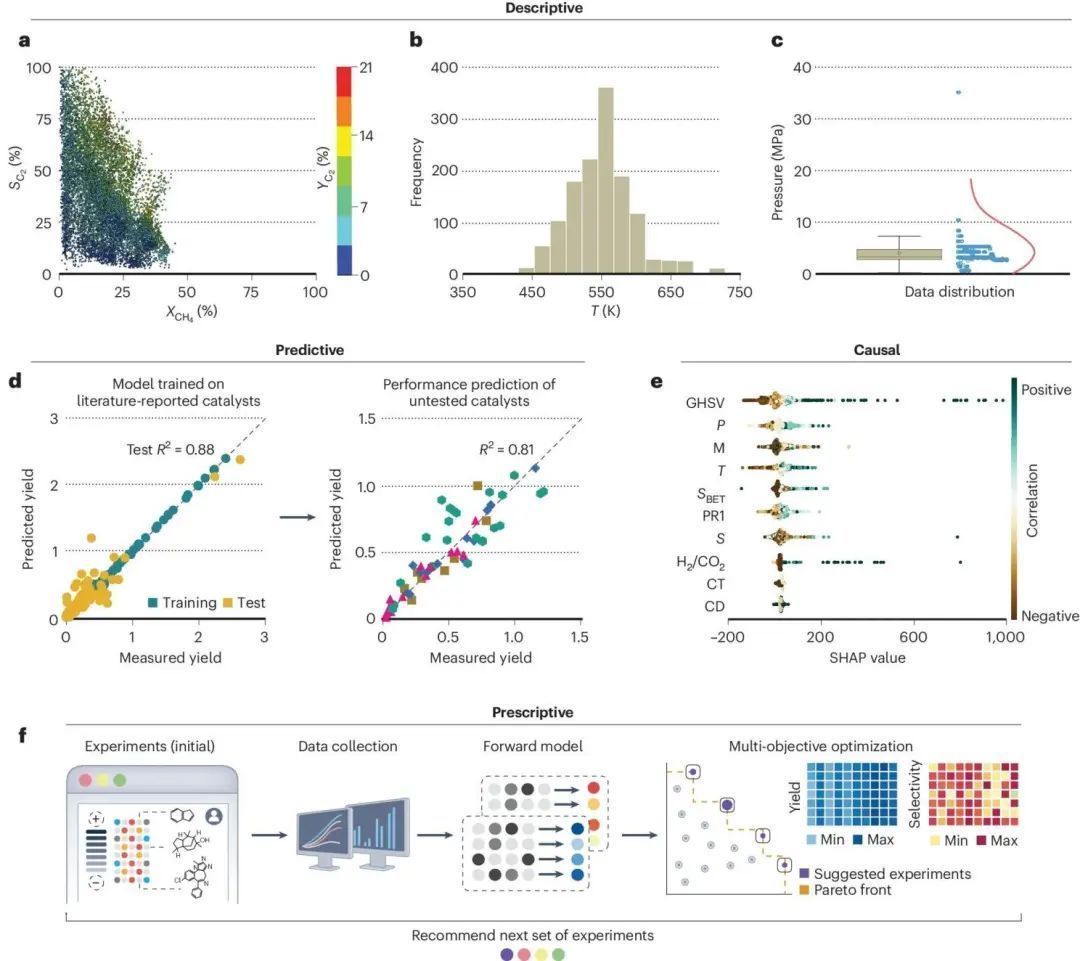
数据科学的基本思想是,数据可以为手头的工作提供见解,而无需过度依赖预测模型。对数据进行统计分析以识别模式和关系,不受理论偏见的影响,被称为描述性分析。对于大型数据集而言,采用可视化技术对于增强描述性推断至关重要。例如,网络图,用于定量分析所使用的不同类型的活性相、促进剂和支持物,以及它们之间的相互作用频率;连续变量(如反应条件)的直方图或小提琴图,用于了解数据分布;以及选择性与转化率映射的二维定位图,用于产生第三维度(图7a-c)。至于催化剂筛选,目前多是遵循试错法,从数据科学的角度来看,催化剂筛选类似于模式匹配,因此非常适合开发以化学为依据的预测分析。根据初步筛选获得的现有数据,预测分析法可将性能与描述符和反应条件相关联,提供内推的可能性,在某些情况下还可外推模型,以确定未经测试的催化剂的性能(图7d),实现节省时间和资源,并确保了更大覆盖面。
虽然经典的ML算法具有更强的预测能力,但它们通常是具有黑箱性质,对相关催化系统的合理化作用微乎其微。而因果分析是通过不断迭代的可解释ML方法,其能够很好地解释催化系统。该方法与模型无关,只根据输入变量对目标变量的贡献(图7e)按其重要性排序。此外,还可以结合实验验证的数据用于模型的再训练,以改进目标变量预测,并提出更可靠的实验改进方案(图7f)。
对于未来,将深度学习算法与先进表征技术相结合,将使实验人员能够以更快的速度揭示催化材料的复杂性,并具有预测能力。与人工图像分析相比,此类算法能减少人为偏差,并提升效率约2000倍(图8a)。而XAFS和XANES产生的复杂数据集对传统分析方法提出了挑战,在此类海量数据集上训练的深度学习算法可以破译活性物种局部环境的定量结构信息,包括它们与吸附物的相互作用,并揭示复杂的反应动力学(图8b)。

文献信息
Suvarna, M., & Pérez-Ramírez, J. (2024). Embracing data science in catalysis research. Nature Catalysis, 1-12.
