
自然光合作用利用太阳能,通过基于分子酶和辅酶的全天候光/暗反应,将水和大气中的CO2转化为碳水化合物,激发了人工光合作用的广泛发展。然而,开发不含贵金属的高效人工光合系统,以及在分子水平上将功能单元合理整合到单一系统中,仍然具有挑战性。
安徽师范大学熊宇杰教授、陆洲教授,利莫瑞克大学Mei-Yan Gao等人报道了一个人工系统,即Cu6团簇和Co三吡啶配合物的组装系统,通过在Cu6团簇上精确整合纳米酶配合物和辅酶Q来模拟自然光合作用。该仿生体系在光反应中有效地将CO2还原为CO,产率达到740.7 μmol·g-1·h-1,并具有至少188小时的高耐久性。
值得注意的是,该系统实现了光和暗反应的解耦,利用酚衍生辅酶Q作为电子存储。通过调节辅酶Q的稳定剂,暗反应时间可延长至8.5小时,完全满足自然昼夜循环要求。该发现促进了人工系统的分子设计,这些系统可以复制自然光合作用的综合功能。 相关工作以《A cluster-nanozyme-coenzyme system mimicking natural photosynthesis for CO2 reduction under intermittent light irradiation》为题在《Nature Communications》上发表论文。这也是熊宇杰教授在《Nature Communications》上发表的第19篇论文。


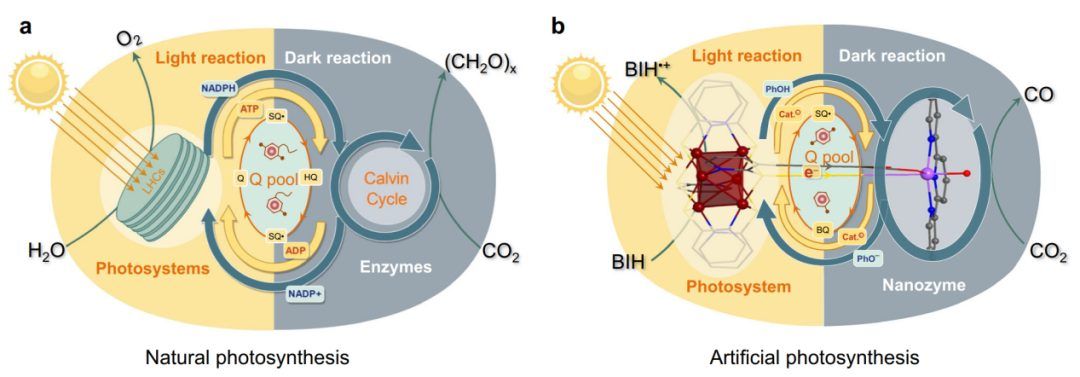
在自然光合作用中(图1a),许多LHC(叶绿素和/或类胡萝卜素色素)收集太阳光来驱动水氧化,产生质子和电子。然后,在辅酶因子的帮助下,电子被有效地储存和转移,从而使特定酶催化剂还原CO2的暗反应得以发生。这些单元之间的密切合作形成了一个高效的系统,在这个系统中,明暗反应的巧妙分离使植物能够在间歇性的阳光下生存。因此,复制这样一个精致系统的关键在于开发相应的功能材料并将它们巧妙地整合在一起。
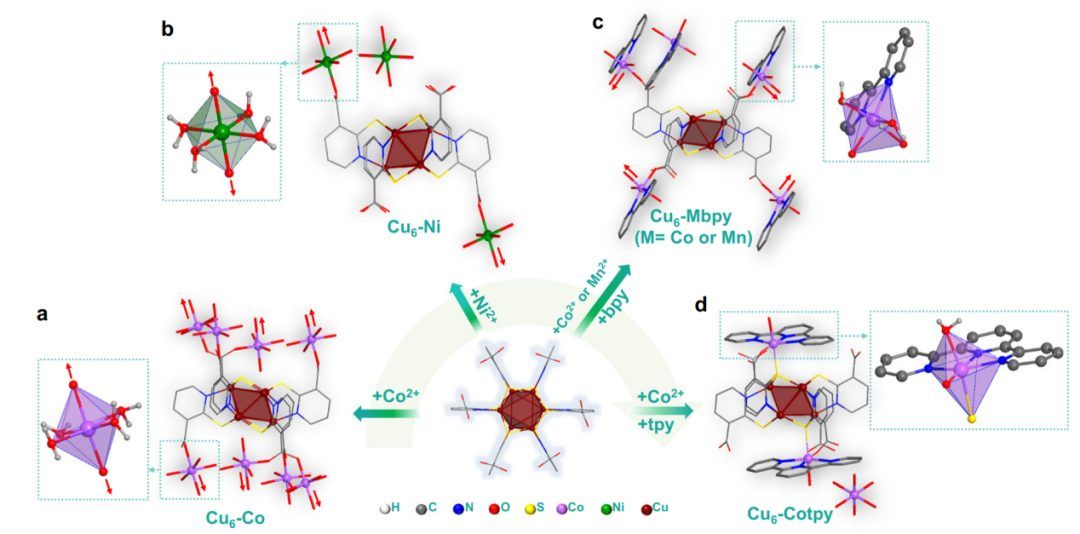
如图2a所示,将裸CoII盐加入到Cu6簇的光合系统中,得到(Cu6(mna)6[Co(H2O)4]3·8H2O的化合物(以下简称Cu6-Co)。[Cu6(mna)6]6-单元中的6个羧酸位点与CoII离子配位,形成一个二维(2D)框架,显示出与4个配位水分子的八面体配位环境。利用Cu6簇的开放位点,可以很容易地在簇上组装不同的金属阳离子。以NiII盐取代CoII盐,得到Cu6-Ni。
为了改变催化中心的配位环境,引入了双齿螯合配体2,2'-联吡啶(bpy)作为修饰剂,考虑到其多种配位模式和多吡啶配合物良好的光还原活性。将bpy引入到Cu6-Co体系中,得到了Cu6-Cobpy(图2c)。
与Cu6-Co化合物相比,两个CoII中心的配位水分子被一个bpy配体取代,bpy配体也起到了末端配体的作用,在一定程度上阻止了结构的延伸。Cu6-Cobpy化合物中存在两种CoII离子,均采用畸变八面体配位环境。一种是通过与两个水分子、mna连接体中的两个O原子和bpy配体中的两个N原子配位形成的。
另一种类型,通过与四个水分子和两个配体中的N原子配合,作为反离子。 为了进一步调整纳米酶的配位环境,采用了三齿螯合配体2,2':6',2'-三吡啶(tpy)。结果得到了Cu6-Cotpy(图2d)。与Cu6-Co相比,三个与Co2+配位的水分子被一个tpy配体取代,作为末端配体,进一步阻止了结构的延伸。
具体而言,Cu6-Cotpy中的CoII离子仍然采用与Cu6-Cobpy一样的扭曲八面体配位环境,并与1个配位水分子、mna连接体中的1个O原子、1个S原子和tpy配体中的3个N原子配位。值得注意的是,在Cu6-Cotpy中,Cu6簇的一个S原子直接与Co中心键合,这与上述化合物明显不同。
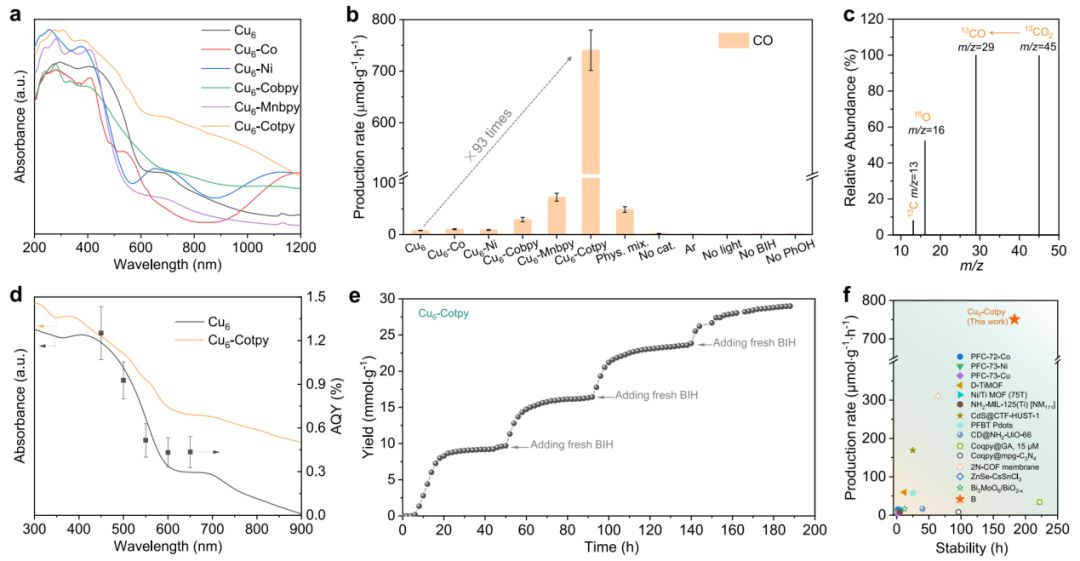
根据其组成和结构信息,通过紫外-可见(UV-Vis)吸收光谱对其光捕获能力进行了评价。如图3a所示,裸Cu6簇表现出相当大的可见光吸收,最高可达约700 nm,其最低未占据分子轨道(LUMO)能级比CO2的还原电位更负,表明所选择的Cu6簇能有效吸收光用于CO2光还原。
另外五种合成的化合物也表现出良好的可见光响应,这可归因于Cu6簇的吸光能力。Cu6-Co和Cu6-Ni在约1100 nm处有明显的d-d激发,表明它们具有单金属位点的特征。在Cu6-Cotpy上实现了最广泛的集光特性,它几乎覆盖了整个太阳光谱,这意味着可以预期更有效的光催化性能。
考虑到纳米酶具有良好的光收集能力和开放金属位点的存在,评估了可见光驱动(λ≥420 nm)的CO2还原性能,无需额外的光敏剂和助催化剂。将10 mg光催化剂超声分散于100 mL CO2饱和的ACN溶液中,ACN溶液中分别含有0.05 M BIH和0.03 M PhOH作为电子供体和质子源。
如图3b所示,原始Cu6簇对CO(8.0 μmol·g-1·h-1)和CH4(0.5 μmol·g-1·h-1)的生成活性可以忽略不计,表明裸Cu6簇对CO2的活化几乎是惰性的。用Co单金属位点修饰团簇(Cu6-Co)后,CO (10.4 μmol·g-1·h-1)和CH4(1.0 μmol·g-1·h-1)的产率略有提高。当Co金属位点被Ni取代(Cu6-Ni)时,也出现了同样的情况,说明水合的单一金属位点对促进CO2活化无效。
当引入了多吡啶配体来调节活性中心的配位环境,如图3b所示,Cu6-Cobpy的CO生成活性提高到29.1 μmol·g-1·h-1,是原始Cu6簇的3.6倍,并且通过羰基化取代配位的H2O可以进一步提高其活性。受到这一改进的鼓舞,进一步将多吡啶配体改为三吡啶。Cu6-Cobpy的CO生成速率为740.7 μmol·g-1·h-1,是裸Cu6簇的93倍。
值得一提的是,Cu6-Cotpy需要很长的预激活时间(5到40小时,在此期间可以检测到可忽略不计的CO),才能获得稳定的性能。这一特征可能归因于活性位点周围局部微环境的潜在重构。 在13C标记的CO2(13CO2)气氛下进行同位素标记实验,在m/z=29(13CO)处检测到明显的信号峰(图3c),证实生成的CO来源于CO2的光还原。
图3d表明,AQY趋势与Cu6的吸收谱吻合较好,而与Cu6-Cotpy的吸收谱不吻合,表明该反应确实是由Cu6的光生电子驱动的。因此,Cu6簇就像自然光合作用中的LHC一样充当光吸收材料。作者还测试了Cu6-Cotpy在四个连续循环中的耐久性,每个循环需要47小时。由于Cu6-Cotpy在乙腈(ACN)中的溶解度较低,每个新循环都通过添加新鲜BIH和用纯CO2更新反应气氛来进行测试。
如图3e所示,Cu6-Cotpy催化CO2转化至少可以保持188 h的活性,表明其具有良好的光稳定性。
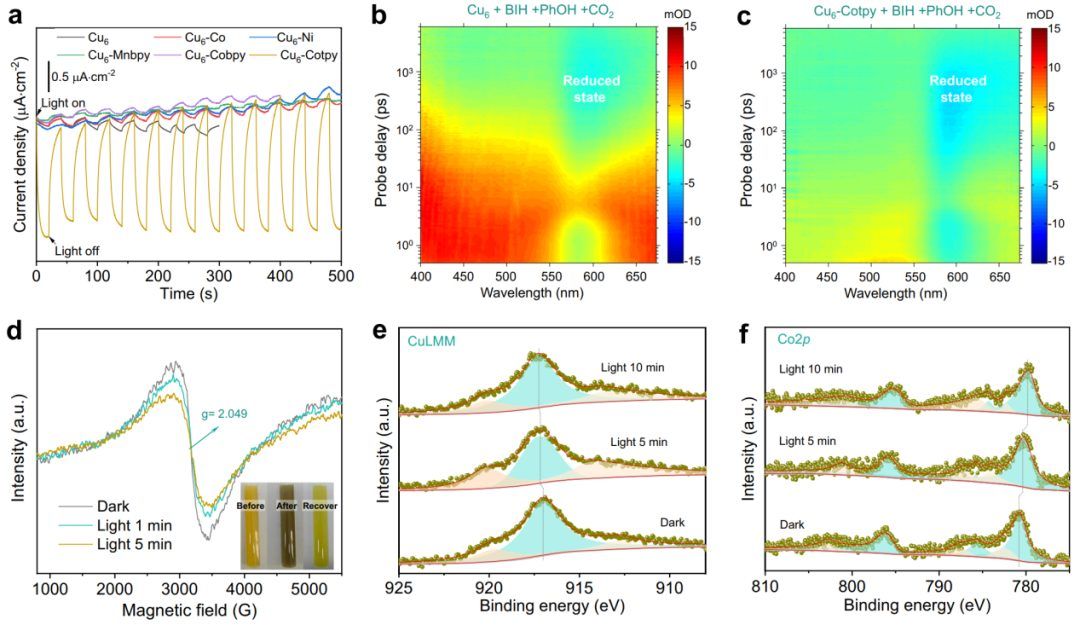
作者首先关注Cu6簇(光收集器)和组装的催化中心(纳米酶)之间的电荷转移性能。如图4a所示,Cu6-Cotpy的瞬态光电流响应明显高于其他五种对应物,表明其具有明显的光生电荷分离能力。
从Cu6-Cotpy的结构来看,这一优势主要体现在两个方面:(1)CoII离子直接与Cu6簇的S键结合,形成了较短的Cu6中心到Co中心的电荷转移通道;(2)tpy配体的大π共轭可以作为储存光生电子的储存器,从而防止Cotpy上的电子回流到Cu6。 在超快时间尺度上,引入PhOH后Cu6的电荷动力学行为明显不同于单独Cu6和BIH存在时Cu6的电荷动力学行为,这表明PhOH也被预结合到Cu6簇的表面。
更重要的是,在PhOH和CO2的存在下,这五种化合物之间的差异变得非常明显。如图4b、c二维TA光谱伪彩色图所示,在~590 nm处Cu6(Cu6-)的还原态引起的负ΔOD信号仅在原始Cu6簇或Cu6-Cotpy中观察到,而在具有1D和2D结构的簇中几乎可以忽略,这表明具有开放位点的簇更有利于氧化BIH生成Cu6-。
基于光激发Cu6到组装的Cotpy的电荷转移动力学信息,进一步致力于确定Cotpy上电子受体的特定位置。首次采用原位ESR光谱,通过检测金属位点在捕获电子前后的自旋状态来分析价态的变化。
如图4d所示,在光照射前,在g=2.049处观察到信号,考虑到该体系中CuI的金属是非磁性的,这可以归因于CoII的高自旋态。可见光照射后,信号强度大大减弱,表明光诱导价态从高自旋态的CoII转变为低自旋态的CoI。这些结果充分表明,Cu6的接受电子被捕获在Co位点上,而Co位点又应作为后续CO2还原的活性位点。 原位XPS光谱进一步证实了这一结论。从高分辨率XPS光谱(图4e)可以看出,随着辐照时间的增加,Cu LMM的峰逐渐向结合能较高的方向移动,说明Cu在光激发下的电子密度降低了。
相比之下,Co 2p的峰值在辐照5 min和10 min后分别向相反方向偏移了约0.4 eV和0.9 eV(图4f),表明Co原子的电子密度随着辐照时间的延长而升高。这些结果表明,Cu6簇的光生电子被转移到Cotpy配合物的Co位点上。

在获得光收集器和活性位点之间的电荷转移行为后,将注意力转向纳米酶的催化机制。采用原位DRIFTS研究了Co位点在受激光催化反应条件下配位环境的动态演变。
如图5a所示,对于未光照的样品,辐照15 min后出现了以668、570和555 cm-1为中心的峰,这可以归因于Cotpy配合物或[Co(H2O)6]2+离子的νCo-O振动吸收。这些峰的出现表明相应的配位键在光照射后发生了变化,当高配位的CoII中心被光生电子和/或反离子与含氧物质(如酚衍生物)配位还原到低配位的CoI位点时,配位键被拉长或减弱。 当辐照时间达到55 min时,在542 cm-1处观察到新的νCo-N的振动吸收,这可归因于溶剂ACN分子与Cotpy配合物的Co中心的配位。随着辐照时间的增加,νCo-N的振动吸收峰强度逐渐增强,νCo-O的振动吸收峰强度逐渐减弱,表明原先与氧配位的Co位点已经转变为与氮配位。
在120 min后,555 cm-1处的峰也消失了,这可能是由于配位的H2O分子完全被ACN取代或Co中心与羧基之间的Co-O键断裂所致。这个漫长的过程应该是光催化反应初始阶段催化剂的预活化时间。 为了解码CO2在动态纳米酶位点上的活化机制,还进行了依赖于辐照时间的近红外原位DRIFT测量。
如图5b所示,照射3min后,在1408 cm-1处首次检测到明显的吸收峰,这是由于*HCO3-的对称拉伸。随着辐照时间的增加,*HCO3-的特征峰逐渐向更高的波数蓝移,同时还观察到其他单齿碳酸盐和双齿碳酸盐的中间态,表明*HCO3-对Co位点的吸附强度增强,*HCO3-进一步通过去质子作用演化为被吸附的碳离子。同时,辐照4min后,在1602 cm-1处出现了将CO2还原为CO的关键中间体*COOH。
总的来说,基于上述原位DRIFTS结果,可以提出CO2的活化机制。CO2最初在活性位点与H2O共吸附形成*HCO3-中间体,然后通过去质子作用演变成*CO32-。在接下来的步骤中,*CO32-接受光生电子,并在照射过程中与质子偶联形成*COOH。
最后,*COOH中间体去质子化形成CO分子。 基于电荷动力学、配位环境演化和中间体物种的信息,利用DFT计算进一步描述了纳米酶的反应途径,并利用其良好定义的结构进行理论建模。Co位点接受两个电子的第一步是Cu6-Cobpy和Cu6-Cotpy上CO2-to-CO还原反应的速率决定步骤(RDS)(图5c),证实了预活化过程对后续催化反应至关重要。Cu6-Cotpy上大多数反应步骤的生成能明显低于Cu6-Cobpy上的生成能,这说明Cu6-Cotpy具有优越的光催化性能。
综上所述,配位结构、电荷动力学、原位、光谱表征和理论计算提供了一个明确的反应途径,如图5d所示。
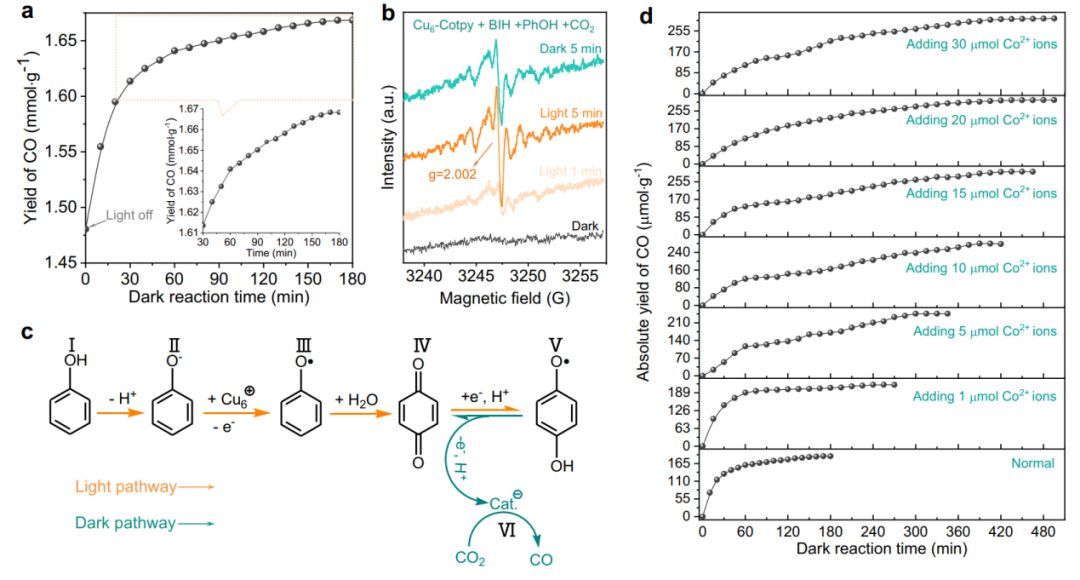
受醌类化合物在自然光合作用中的电子穿梭和储存作用的启发,作者选择苯酚来模拟这一功能。为了证明这一概念,作者评估了Cu6-Cotpy在可见光照射2小时后的黑暗处理中,PhOH作为苯醌(BQ)的前体存在下的催化性能。
如图6a所示,在关闭入射光后,CO的析出反应至少可以持续3小时。析出速率与存储电子数呈一级动力学关系,表明存储电子数是暗反应的直接驱动力。
此外,在进一步光照下,Cu6-Cotpy催化剂可以回收用于暗反应,表明成功复制了光反应与暗反应解耦的功能。 此外,作者还研究了辐照时间与CO产率之间的关系。如图6c所示,当暗反应时间从100 min延长至180 min时,将光照时间从1 h延长至2 h, CO的绝对产率从121.4 μmol·g-1提高到188.9 μmol·g-1,表明存储电子的数量与预照射时间有关。然而,当辐照时间延长至2 h以上时,CO产量的增长停滞不前,说明在照射2 h后达到了储存饱和。
因此,可以得出结论,光反应的高活性是暗反应进行的先决条件。 催化性能和光谱表征共同阐明了暗反应的机理。
如图6c所示,在光反应中,PhOH被去质子化形成苯酚阴离子(II),然后被光生成的空穴氧化为苯酚自由基(III)。在此过程中,PhOH作为双功能电子和质子供体,有利于光反应,而不是直接加入醌类作为电子储存器。随后,由于酚自由基非常活跃,它们会从富氧物质(如水)中捕获氧气,形成BQ(IV)。BQ随后被过量的光生电子还原,形成SQ自由基(V)。
事实上,由于SQ自由基寿命短,不能被隔离,但可以通过与一个或多个金属中心配合来稳定。在该体系中,SQ自由基很可能与CoII的反离子配位。结果也证实了这一特点,与Cu6-Cotpy的正常反应体系相比,在光反应体系中加入额外的CoII离子可以进一步延长暗反应时间(图6d)。
综上所述,本文通过将纳米酶复合物和辅酶Q组装在原子精确的Cu6簇上,构建了一个人工光合系统Cu6-Cotpy,以模拟自然光合作用的光/暗反应过程。在该设计中,通过优化纳米酶的金属中心和外围配体,精心调整了纳米酶的配位环境。结果表明,以三吡啶钴络合物(Cotpy)为纳米酶,实现了高效的CO2光还原。Cu6-Cotpy的光还原速率为740.7 μmol·g-1·h-1,CO生成的选择性高达99%,并且具有至少188 h的高耐久性。这使其成为迄今为止报道的最有效、最耐用的团簇基光催化剂。
文献信息
A cluster-nanozyme-coenzyme system mimicking natural photosynthesis for CO2 reduction under intermittent light irradiation,Nature Communications,2024. https://www.nature.com/articles/s41467-024-53377-0
