为扩大工作电压,人们应用计算化学来寻找具有较低或较高氧化还原电位的有机分子(氧化还原电位和酸度常数决定了质子和电子的热力学驱动力,可用于筛选和设计具有特定功能的新材料)。虽然基于第一性原理分子动力学模拟可以准确描述电解液的溶剂化结构,但仍面临着计算成本和计算精度等多方面的限制。
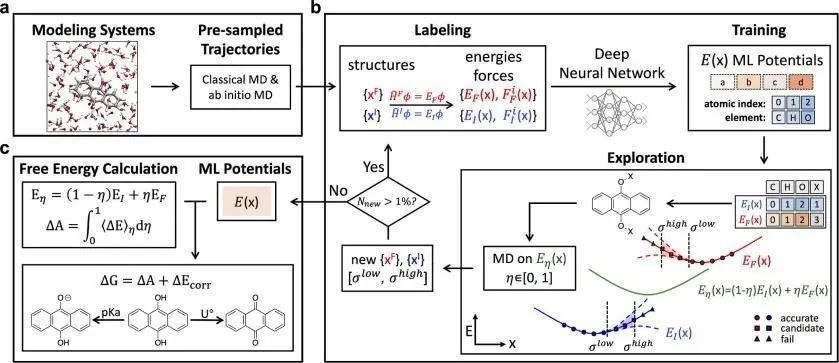
厦门大学/人工智能应用电化学联合实验室(AI4EC Lab)程俊等人开发了一种自动化工作流程,构建了一种通用的机器学习势函数(MLP),从而高效地计算具有氧化还原作用的有机分子的氧化还原电位或酸度常数。 在设计过程中,作者沿着热力学反应路径为MLP收集和更新训练数据集,确保了从初始状态到最终状态的准确预测,同时为热力学性质计算奠定良好基础。
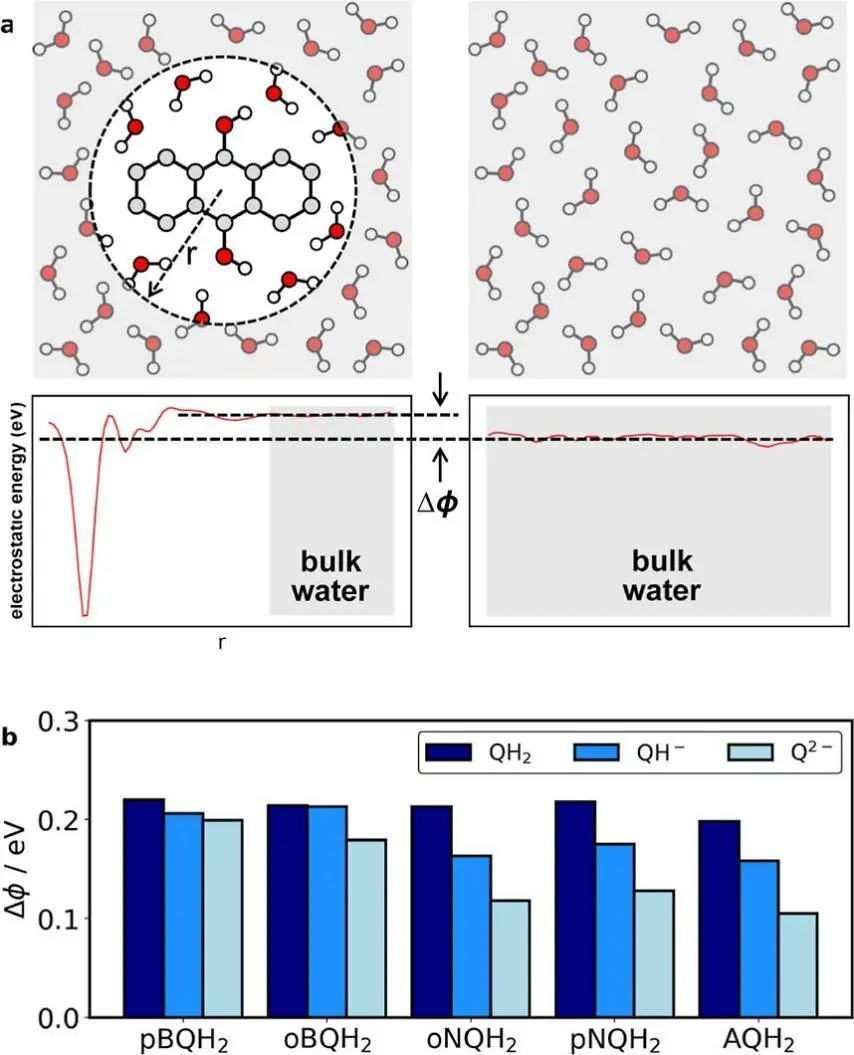
作者希望使用较小的数据集来训练精确的MLP,所以在工作流程设计中使用了杂化泛函以获得更好的氧化还原电位预测精度。此外,该工作流程还可扩展到更高层次的电子结构理论,如无规相近似和基于波函数的量子化学。同时,该工作流程综合了先前研究中被忽视的溶剂化环境,为真实环境中有机分子的RFB进行高通量筛选创造了新的可能性。
对于后续研究,作者所设计的工作流程还可以与具有长程静电相互作用的MLP进一步结合,进一步提高自由能计算的精度。